
引言
晶格常数是晶体结构的核心基础参数,直接决定晶体的几何构型、原子间相互作用及宏观物理化学性质,其精准计算是材料模拟与性能预测的前提的第一步。
下面为大家详细介绍在鸢算科技HPC集群上,如何利用VASP软件针对二维材料(六方晶胞石墨烯)和三维材料(立方晶胞Cu₂O)进行晶格常数测试。
测试材料参数
本次测试选取两种典型晶系材料,覆盖二维、三维不同计算场景,方便大家全面掌握操作要点,具体参数如下:
1. 石墨烯:属于二维六方结构,晶胞参数满足a=b≠c,α=β=90°,γ=120°,其晶格常数实验值约为2.46 Å。
2. 氧化亚铜(Cu₂O):属于典型立方晶系,晶胞参数满足a=b=c,α=β=γ=90°,在Materials Studio软件中提供的标准晶格常数值为4.27 Å。
测试方法
1. 手动调整法
通过不断修改POSCAR文件中的晶格常数数值,分别提交计算任务并获取对应能量,绘制能量-晶格常数曲线,曲线最低点对应的即为最优晶格常数值,适合需要精准控制优化过程的场景。
2. 直接优化法
通过设置INCAR文件中ISIF=3参数,让软件自动对晶胞的晶格常数和原子位置进行同步优化,无需手动调整,操作高效便捷,适合快速获取精准晶格常数。
测试过程
手动调整法——石墨烯
以不同的晶格常数作为文件夹的名字创建新的文件夹
mkdir 2.16 2.26 2.36 2.46 2.56 2.66 2.76在各个文件夹内准备VASP运行的基本输入文件,注意POSCAR里的晶格常数需要手动调整
输入文件——POSCAR
Graphene
1.0
a 0 0
-a/2 √3a/2 0
0 0 20
C
2
Direct
0.666666666 0.333333333 0.5
0.333333333 0.666666666 0.5在不同的文件夹内,依次设置a的值为2.16、2.26、2.36、2.46、2.56、2.66和2.76Å
输入文件——INCAR
IBRION = 2
ISIF = 2
NSW = 20
EDIFFG = -0.01
POTIM = 0.5
ISTART = 0
ICHARG = 2
LREAL = .FALSE.
ENCUT = 520
ISMEAR = 0
SIGMA = 0.05
EDIFF = 1E-6
NELM = 60
NELMIN = 2
LWAVE = .FALSE.
LCHARG = .FALSE.输入文件——KPOINTS
K-POINTS
0
Gamma
7 7 1
0 0 0输入文件——POTCAR
在有POSCAR的文件夹内,使用以下命令直接生成POTCAR文件
potcar提交脚本
准备输入提交计算的脚本,不同集群下的脚本内容有差异,可以联系集群管理员获取
cp /public/slurm_scripts/vasp/gpu/vasp.6.5.1/sub_vasp.sh ./统计结果(a=2.16-2.76)
计算完成后,在每个文件夹使用下面的命令抓取能量
awk '/E0/ {last=$5} END {print last}' OSZICAR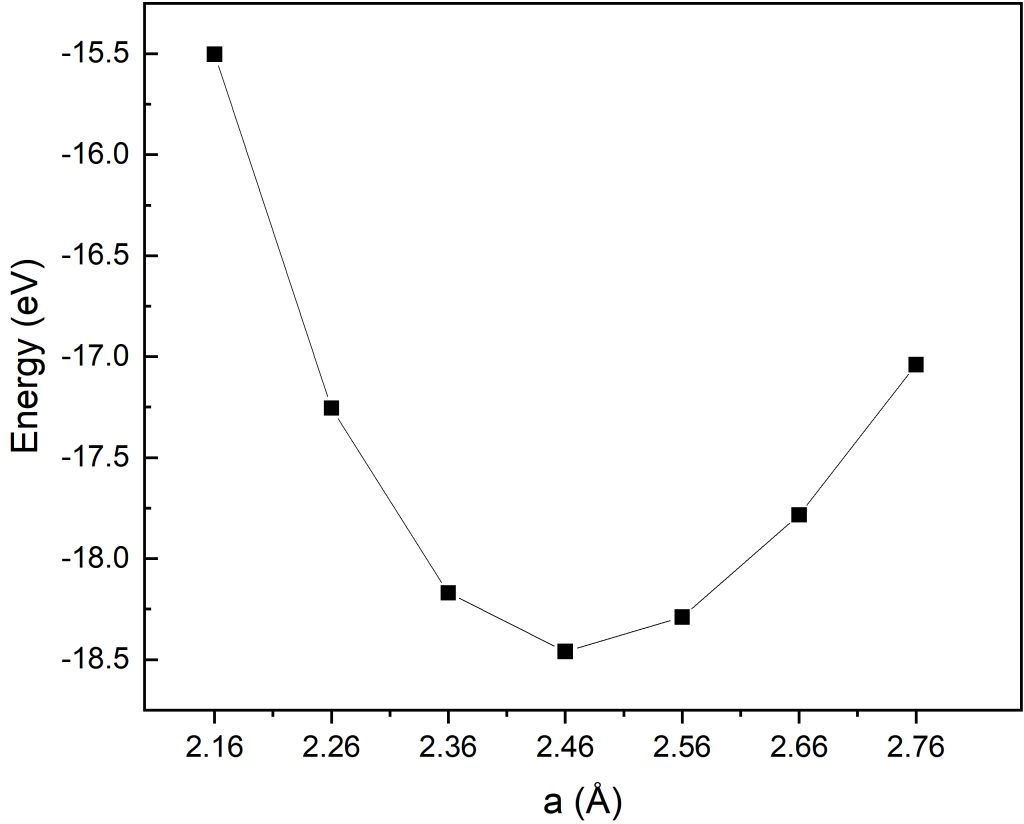
首先对晶格常数a=2.16-2.76进行优化计算,通过不同晶格常数下的体系总能对比分析,由上图结果可明显看出:在该取值范围内,晶格常数a=2.46时体系总能达到最低,由此可以锁定最优晶格常数落在(2.36-2.56)窄区间内。
统计结果(a=2.36-2.56)
为进一步提升晶格常数的收敛精度,在初筛最优值附近设置加密取值区间(2.36-2.56)。
和上述同样的方法,以不同的晶格常数作为文件夹的名字创建新的文件夹
for a in $(seq 2.36 0.01 2.56); do mkdir $a; doneINCAR、KPOINTS和POTCAR文件和上述保持相同,排除输入参数对结果的干扰,仅对晶格常数进行小步长精细扫描。
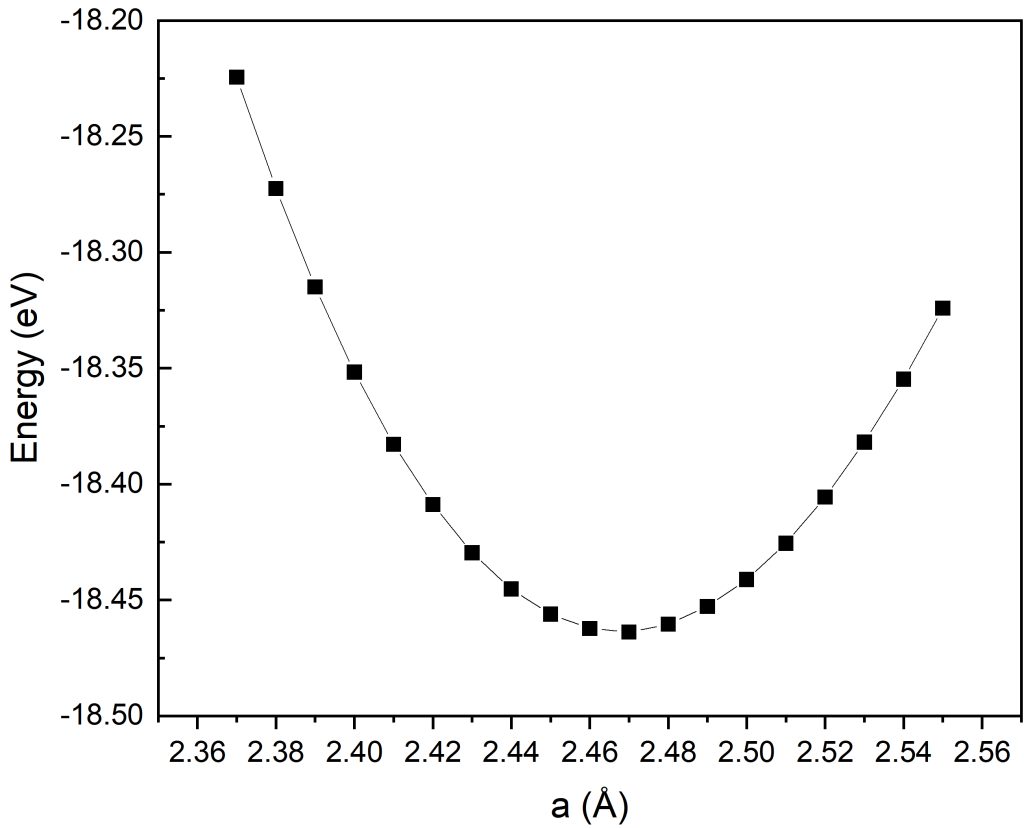
对精细区间的能量拟合结果分析可得:体系总能随晶格常数先降低后升高,呈现典型抛物线变化规律,在a=2.47Å处体系总能取得全局极小值。
综合粗扫初筛与精细加密两轮优化计算结果,最终确定本计算模型下石墨烯的最优晶格常数为2.47Å。
直接优化法——石墨烯
直接优化法不需要手动调整POSCAR中的晶格常数,而是在INCAR中直接设置ISIF=3,一次结构弛豫计算直接得到优化好的晶格常数。
对于二维材料的直接优化,需要使用特殊编译后可以固定晶轴优化的VASP版本(https://github.com/Chengcheng-Xiao/VASP_OPT_AXIS),同时也要在INCAR中添加固定晶轴优化的相关参数IOPTCELL
鸢算科技的集群为客户提供固定晶轴版本的VASP的编译,需要使用的联系集群管理员即可
输入文件——POSCAR
Graphene
1.0
2.46 0 0
-1.23 2.130422493309719 0
0 0 20
C
2
Direct
0.666666666 0.333333333 0.5
0.333333333 0.666666666 0.5输入文件——INCAR
IBRION = 2
ISIF = 3
IOPTCELL = 1 1 0 1 1 0 0 0 0
NSW = 20
EDIFFG = -0.01
POTIM = 0.5
ISTART = 0
ICHARG = 2
LREAL = .FALSE.
ENCUT = 520
ISMEAR = 0
SIGMA = 0.05
EDIFF = 1E-6
NELM = 60
NELMIN = 2
LWAVE = .FALSE.
LCHARG = .FALSE.输入文件——KPOINTS
K-POINTS
0
Gamma
7 7 1
0 0 0输入文件——POTCAR
在有POSCAR的文件夹内,使用以下命令直接生成POTCAR文件
potcar提交脚本
准备输入提交计算的脚本,不同集群下的脚本内容有差异,可以联系集群管理员获取
cp /public/slurm_scripts/vasp/gpu/vasp.6.5.1/sub_vasp_optcell.sh ./优化结果
计算结束后直接查看CONTCAR中的晶格常数矩阵
Graphene
1.000000000000000
2.4678323832750126 0.0000000000000000 0.0000000000000000
-1.2339219000591615 2.1372022404392768 0.0000000000000000
0.0000000000000000 0.0000000000000000 20.0000000000000000
C
2
Direct
0.6666626750572725 0.3333273249427243 0.5000000000000000
0.3333273249427243 0.6666626750572725 0.5000000000000000因此直接优化法得到的晶格常数是2.468Å
手动调整法——氧化亚铜
以不同的晶格常数作为文件夹的名字创建新的文件夹
mkdir 2.16 2.26 2.36 2.46 2.56 2.66 2.76在各个文件夹内准备VASP运行的基本输入文件,注意POSCAR里的晶格常数需要手动调整
输入文件——POSCAR
Cu2O
1.0
$a 0 0
0 $a 0
0 0 $a
Cu O
4 2
Direct
0.250000000 0.250000000 0.250000000
0.750000000 0.750000000 0.250000000
0.750000000 0.250000000 0.750000000
0.250000000 0.750000000 0.750000000
0.000000000 0.000000000 0.000000000
0.500000000 0.500000000 0.500000000在不同的文件夹内,依次设置a的值为4.07、4.17、4.27、4.37和4.47Å
输入文件——INCAR
IBRION = 2
ISIF = 2
MAGMOM = 4*3 2*0
ISPIN = 2
NSW = 200
EDIFFG = -0.01
POTIM = 0.5
ISTART = 0
ICHARG = 2
LREAL = .FALSE.
ENCUT = 520
ISMEAR = 0
SIGMA = 0.05
EDIFF = 1E-6
LWAVE = .FALSE.
LCHARG = .FALSE.输入文件——KPOINTS
K-POINTS
0
Gamma
4 4 4
0 0 0输入文件——POTCAR
在有POSCAR的文件夹内,使用以下命令直接生成POTCAR文件
potcar提交脚本
准备输入提交计算的脚本,不同集群下的脚本内容有差异,可以联系集群管理员获取
cp /public/slurm_scripts/vasp/gpu/vasp.6.5.1/sub_vasp.sh ./统计结果(a=4.07-4.47)
计算完成后,在每个文件夹使用下面的命令抓取能量
awk '/E0/ {last=$5} END {print last}' OSZICAR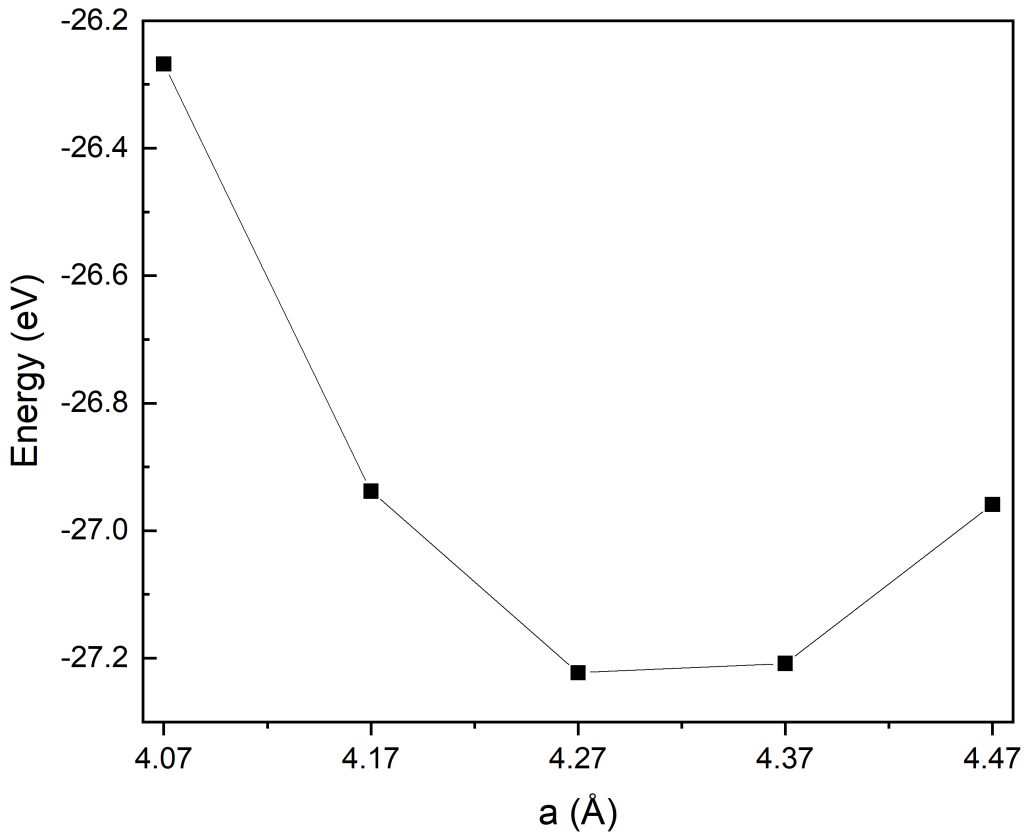
首先对晶格常数a=4.07-4.47进行优化计算,通过不同晶格常数下的体系总能对比分析,由上图结果可明显看出:在该取值范围内,晶格常数a=4.27时体系总能达到最低,由此可以锁定最优晶格常数落在(4.17-4.37)窄区间内。
统计结果(a=4.17-4.37)
为进一步提升晶格常数的收敛精度,在初筛最优值附近设置加密取值区间(2.36-2.56)。
和上述同样的方法,以不同的晶格常数作为文件夹的名字创建新的文件夹
for a in $(seq 4.18 0.01 4.36); do mkdir $a; doneINCAR、KPOINTS和POTCAR文件和上述保持相同,排除输入参数对结果的干扰,仅对晶格常数进行小步长精细扫描。
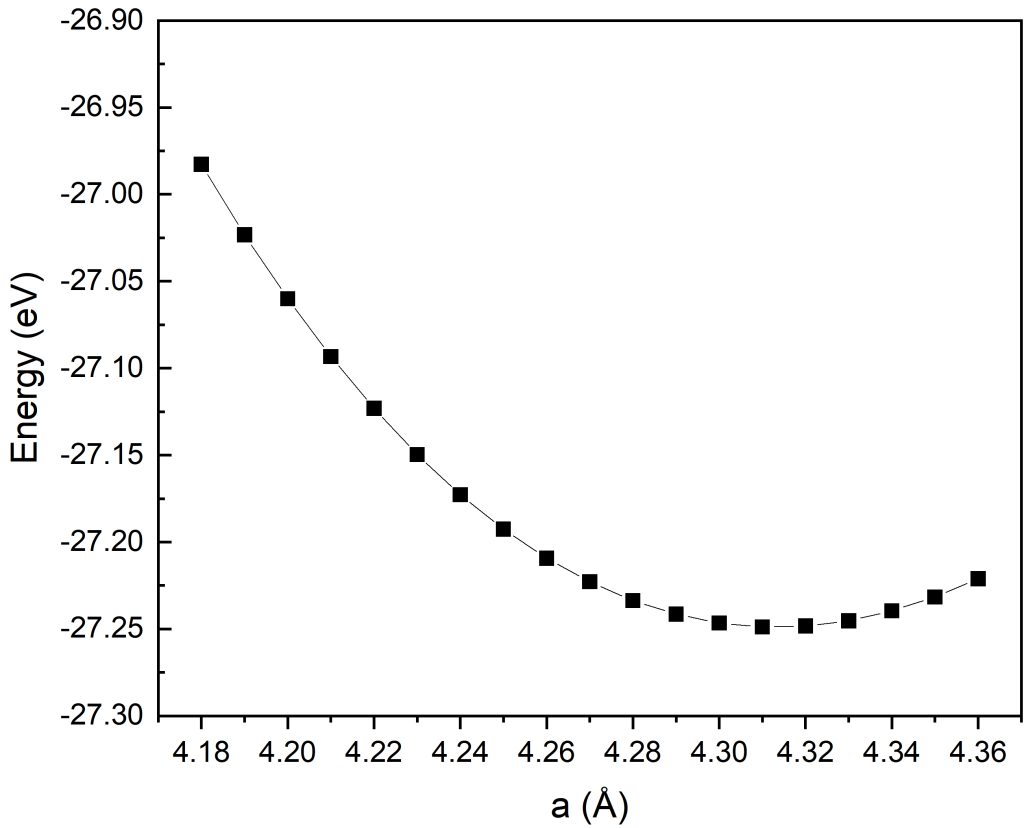
对精细区间的能量拟合结果分析可得:体系总能随晶格常数先降低后升高,呈现典型抛物线变化规律,在a=4.31Å处体系总能取得全局极小值。
综合粗扫初筛与精细加密两轮优化计算结果,最终确定本计算模型下氧化亚铜的最优晶格常数为4.31Å。
直接优化法——氧化亚铜
输入文件——POSCAR
Cu2O
1.0
4.37 0.0000000000 0.0000000000
0.0000000000 4.37 0.0000000000
0.0000000000 0.0000000000 4.37
Cu O
4 2
Direct
0.250000000 0.250000000 0.250000000
0.750000000 0.750000000 0.250000000
0.750000000 0.250000000 0.750000000
0.250000000 0.750000000 0.750000000
0.000000000 0.000000000 0.000000000
0.500000000 0.500000000 0.500000000输入文件——INCAR
IBRION = 2
ISIF = 3
MAGMOM = 4*3 2*0
ISPIN = 2
NSW = 200
EDIFFG = -0.01
POTIM = 0.5
ISTART = 0
ICHARG = 2
LREAL = .FALSE.
ENCUT = 520
ISMEAR = 0
SIGMA = 0.05
EDIFF = 1E-6
LWAVE = .FALSE.
LCHARG = .FALSE.输入文件——KPOINTS
K-POINTS
0
Gamma
4 4 4
0 0 0输入文件——POTCAR
在有POSCAR的文件夹内,使用以下命令直接生成POTCAR文件
potcar提交脚本
准备输入提交计算的脚本,不同集群下的脚本内容有差异,可以联系集群管理员获取
cp /public/slurm_scripts/vasp/gpu/vasp.6.5.1/sub_vasp.sh ./优化结果
计算结束后直接查看CONTCAR中的晶格常数矩阵
Cu2O
1.000000000000000
4.3118049152124875 0.0000000000000000 -0.0000000000000000
0.0000000000000000 4.3118049152124875 0.0000000000000000
-0.0000000000000000 0.0000000000000000 4.3118049152124875
Cu O
4 2
Direct
0.2500000000000000 0.2500000000000000 0.2500000000000000
0.7500000000000000 0.7500000000000000 0.2500000000000000
0.7500000000000000 0.2500000000000000 0.7500000000000000
0.2500000000000000 0.7500000000000000 0.7500000000000000
0.0000000000000000 0.0000000000000000 0.0000000000000000
0.5000000000000000 0.5000000000000000 0.5000000000000000因此直接优化法得到的晶格常数是4.312Å
手动调整法——自动测试脚本
在鸢算科技集群里的使用方法为(假设脚本的名字为lc.sh)
sbatch lc.sh二维材料(六方晶胞)
#!/bin/bash
#SBATCH -o %j.out
#SBATCH -J job_name
#SBATCH -p GPU
#SBATCH --nodes=1
#SBATCH --ntasks=1
#SBATCH --gres=gpu:1
source /public/toolkit/intel/oneapi_2023/mkl/latest/env/vars.sh
module load /public/toolkit/nvidia/hpc_sdk/modulefiles/nvhpc/25.3
export NO_STOP_MESSAGE=1
export OMP_NUM_THREADS=1
export vasp_path=/public/software/vasp/GPU/vasp.6.5.1/vasp_std
sqrt3=1.7320508075
echo > cellparameter
for ii in $(seq 2.37 0.01 2.55); do # 在这里设置要测试的晶格常数的范围和步长
#define lattice parameters: a, b, c:
a=`echo "scale=16; $ii" |bc`
b=`echo "scale=16; -$a/2" |bc`
c=`echo "scale=16; $a/2*$sqrt3" |bc`
mkdir $ii
#在下面的两个EOF包括的内容里填写POSCAR,并把晶格常数a改为$a,$b和$c
cat>POSCAR-$ii<<EOF
graphene
1.0
$a 0 0
$b $c 0
0 0 20
C
2
Direct
0.666666666 0.333333333 0.5
0.333333333 0.666666666 0.5
EOF
mv POSCAR-$ii $ii
cp POTCAR KPOINTS INCAR $ii
cd $ii
cp POSCAR-$ii POSCAR
mpirun -np $SLURM_NTASKS --bind-to none $vasp_path
cd ..
E=`tail -1 ${ii}/OSZICAR` ; echo $a $E >>cellparameter
done
cat cellparameter三维材料(立方晶胞)
#!/bin/bash
#SBATCH -o %j.out
#SBATCH -J job_name
#SBATCH -p GPU
#SBATCH --nodes=1
#SBATCH --ntasks=1
#SBATCH --gres=gpu:1
source /public/toolkit/intel/oneapi_2023/mkl/latest/env/vars.sh
module load /public/toolkit/nvidia/hpc_sdk/modulefiles/nvhpc/25.3
export NO_STOP_MESSAGE=1
export OMP_NUM_THREADS=1
export vasp_path=/public/software/vasp/GPU/vasp.6.5.1/vasp_std
echo > cellparameter
for ii in $(seq 4.18 0.01 4.36); do # 在这里设置要测试的晶格常数的范围和步长
a=`echo "scale=16; $ii" |bc`
mkdir $ii
#在下面的两个EOF包括的内容里填写POSCAR,并把晶格常数a改为$a
cat>POSCAR-$ii<<EOF
Cu2O
1.0
$a 0 0
0 $a 0
0 0 $a
Cu O
4 2
Direct
0.250000000 0.250000000 0.250000000
0.750000000 0.750000000 0.250000000
0.750000000 0.250000000 0.750000000
0.250000000 0.750000000 0.750000000
0.000000000 0.000000000 0.000000000
0.500000000 0.500000000 0.500000000
EOF
mv POSCAR-$ii $ii
cp POTCAR KPOINTS INCAR $ii
cd $ii
cp POSCAR-$ii POSCAR
mpirun -np $SLURM_NTASKS --bind-to none $vasp_path
cd ..
E=`tail -1 ${ii}/OSZICAR` ; echo $a $E >>cellparameter
done
cat cellparameter